Una delle malattie ereditarie più comuni
La fibrosi cistica costituisce una delle malattie ereditarie più frequenti nel mondo occidentale, con una frequenza di 1:2500 nascite. È ereditata in modo autosomico recessivo e la frequenza di portatori si calcola in 1:20-1:25 individui.
Si tratta di una malattia grave che influenza numerosi organi, come l’apparato respiratorio, il pancreas e l’apparato digestivo. Negli uomini, alcune mutazioni possono provocare sterilità (assenza bilaterale di vasi deferenti (CBAVD)).
Mutazioni nella Fibrosi Cistica
Il gene responsabile della malattia è stato identificato nel 1989 ed è chiamato regolatore della conduttanza transmembrana della fibrosi cistica (cystic fibrosis trans membrane regulator, CFTR).
Fino ad oggi, sono state individuate >1600 lesioni (mutazioni) nel materiale genetico dei pazienti affetti da fibrosi cistica, la maggior parte delle quali sono ritenute patologiche e presentano una frequenza che varia geograficamente e per popolazione.
La mutazione genetica più diffusa nel mondo greco, con una frequenza del 53,4%, è la Delta-F508, ritenuta tra le mutazioni più gravi per sintomatologia.
Le mutazioni immediatamente successive per frequenza di apparizione, sul territorio greco, coprendo un altro 17,7% della popolazione, sono:
- 621+1G>T (5,7%)
- G542X (3,9%)
- N1303K (2,6%)
- 2789+5G>A (1,7%)
- 2183AA>G (1,4%)
- E822X (1,4%)
- R1158X (1%)
Le restanti hanno una frequenza inferiore all’1%.
Fibrosi Cistica e Fecondazione
Fibrosi Cistica e Fecondazione
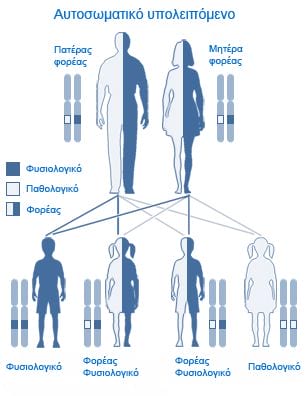
Ogni individuo ha due copie del gene CFTR. Per essere affetto di fibrosi cistica, deve presentare mutazioni in entrambe le copie, cioè deve essere omozigote, mentre chi presenta mutazioni solo in una copia, cioè eterozigote e portatore della malattia.
Un bambino affetto da fibrosi cistica, nasce quando entrambi i genitori presentano mutazioni genetiche, e non quando dette mutazioni interessano uno dei due. Se poi, entrambi i genitori sono portatori, esiste il 25% di possibilità che il bambino nasca con la fibrosi cistica.
Controllo della Fibrosi Cistica
L’unico modo per riconoscere i portatori di fibrosi cistica è lo screening genetico molecolare, vale a dire il rilevamento di mutazioni genetiche nel gene CFTR, a livello dell’intera popolazione. Il controllo per la fibrosi cistica al fine di individuarne i portatori, si effettua:
- nei casi di programmazione familiare
- quando esiste un’anamnesi a livello familiare
- quando si osserva intestino iperecogeno nell’ecografia
- quando si esamina la subfertilità
Nei casi di programmazione familiare, se entrambi i genitori sono portatori, allora le informazioni sul feto possono essere fornite dai metodi di diagnosi preimpianto e controllo prenatale.