Une des maladies héréditaires les plus courantes
La mucoviscidose constitue l’une des maladies héréditaires les plus courantes dans le monde occidental, avec une incidence de 1 sur 2 500 naissances. Elle est transmise selon un mode autosomique récessif et la fréquence des porteurs est estimée à 1 sur 20 – 1 sur 25 personnes.
Il s’agit d’une maladie grave qui affecte de nombreux organes tels que les systèmes respiratoire, digestif et pancréatique. Chez les hommes, certaines mutations peuvent entraîner l’infertilité (absence bilatérale des canaux déférents (ABCD)).
Mutations dues à la fibrose cystique
Identifié en 1989, le gène responsable est nommé CFTR (cystic fibrosis trans membrane regulator). À ce jour, 1.600 défaillances (mutations) ont été détectées dans le matériel génétique des patients atteints de mucoviscidose, dont la plupart sont considérées comme pathologiques, présentant une fréquence qui diffère en fonction des critères géographiques et démographiques.
La mutation la plus fréquente observée en Grèce avec une fréquence de 53,4 % est la mutation ΔF508 qui est considérée comme la plus grave des mutations en termes de symptomatologie.
Les mutations en termes d’incidence en Grèce sont les suivantes: 621+1G>T (5,7%), G542X (3,9%), N1303K (2,6%), 2789+5G>A (1,7%), 2183AA>G (1,4%), E822X (1,4%) et R1158X (1%), ce qui représente 71,1 % de la population grecque.
Les autres présentent une incidence inférieure à 1 %.
Fibrose cystique et fécondation
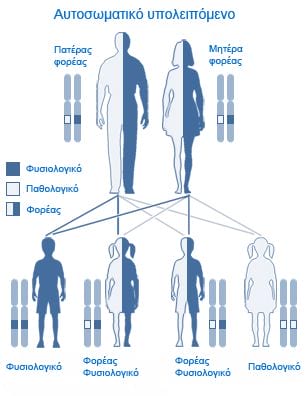
Chaque individu porte deux copies du gène CFTR. Pour être atteint de mucoviscidose, un individu doit être homozygote, c.à.d. présenter une mutation des deux copies. Une personne portant une mutation dans une seule copie est dite hétérozygote et est porteuse de la maladie.
Pour qu’un enfant ne soit pas atteint de mucoviscidose, il faut que les deux parents ne soient pas porteurs de mutation ou qu’un seul des deux soit porteur. Si les deux parents sont porteurs, il existe 25 % de risque qu’un enfant naisse atteint de mucoviscidose.
Contrôle de la fibrose cystique
Le test génétique (le dépistage des mutations du gène CFTR) constitue la seule façon d’identifier les porteurs de mucoviscidose au sein de l’ensemble de la population.
Le dépistage de la mucoviscidose en vue d’identifier des porteurs de la maladie est réalisé en cas de planification familiale, quand il existe des antécédents familiaux, lorsqu’un intestin hyperéchogène est détecté à l’échographie et lors du contrôle de la fertilité.
En cas de planification familiale, si les deux parents sont porteurs, les méthodes de diagnostic préimplantatoire et de dépistage prénatal permettent de donner des informations sur l’embryon.